|
|
In the modelling practice it is not just needed to correctly place one rotamer in an otherwise unmodified protein, but often around half of all side chains need to be altered. That means that in many cases the environment is either incorrect, or which causes slightly less of a problem, not yet complete. This makes the rotamer choice one step mor complicated because even the best force field or scorings function can not overcome the problem of the chicken and the egg. In order to correctly position side chain A, all its neighbours need to be in the perfect position already, but these perfect positions can not be determined before side chain A is correctly modelled.
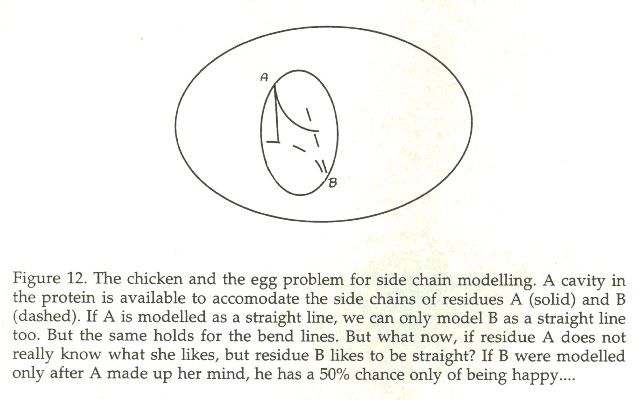
Figure 12 indicates in a simplified fashion what is the problem. If there is limited space to model two side chains, there could be, for example, two seemingly equally good solutions. In such a simple case the quality of the outcome is determined solely by the quality of the force field, or scorings function. Our professional gambler likes this situation, because some understanding of statistics will help. If the number of choices is limited the modeller can simply enumerate them and select the one with the highest probability of being right. If there are more than two side chains whos positions influence each other, it seems that we rapidly run into a combinatorial explosion. The death-end elimination method [101,102] was the first attempt to solve this combinatorial problem without the use of Monte Carlo methods. The rationale of the authors was: łWhy gamble if everything can be calculated˛? Chinea et al., [107] actually studied this problem, and concluded that it is not a problem at all, or at least the problem is not what previously thoughtlessly was assumed that it was.
|
|
If many new side chains have to be placed in the model, multiple side chains can potentially occupy the same space. A big problem for most modeling methods is that they are based on an energy function that includes contacts between residues distant in the sequence, but close in space. This implies that the whole molecule needs to be build before any selected rotamer can be evaluated. This leads to a "chicken and the egg" problem. In order to place the first residue correctly, all other residues should already have been placed correctly. So, where to start? Several techniques have been described to overcome this problem. Monte Carlo procedures [97,100] seem the most apropriate for this purpose, but Desmet et al., [101,102] already indicated that other solutions might exist.
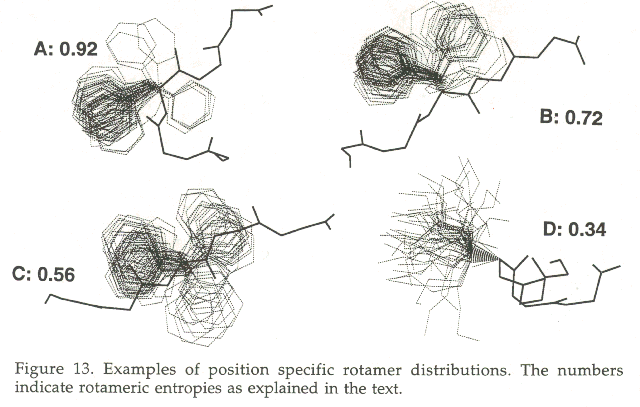
Figure 13 shows several examples of position specific rotamer distributions. Figure 13A shows an example where the position specific rotamer distribution is extremely narrow. If such a case would ocurr in a modeling study, this residue should be modeled immediately, and never looked at again. Figure 13D shows an example of a very wide rotamer distribution. Such a residue should obviously be modeled late in the modeling procedure, because it can much more easily adapt to the space left to it after all other side chains have been placed. The equivalent of figure 13D for our professional gambler is a set of cards that requires a lot of knowledge about the the distribution of cards still to come in order to make the best decision. His chances of earning some money would in such a case greatly enhance if the completion of this game could be postponed till a later stage. And that is what our modeler is allowed to do. If it is not (yet) clear how to model a side chain, she just waits, builds other side chains first and hopes that thereby the number of choices for the problematic side chains are reduced.
Chinea et al., [107] based their modeling strategy on simple probability principles. The narrower the rotamer distribution, the higher the probabilty that this is the rotamer needed in the structure to be modeled. To quantify rotamer distributions a rotameric entropy was defined. In figure 13 some examples of rotamer distributions are shown and the derived rotameric entropies are given. The rotameric entropy is defined by E=(Ptot/P)*(Ftot/F) in which P is the population with c1 within 45 degrees of the most populated of the three standard c1 values (60 degrees, 180 degrees, 300 degrees), Ptot is the sum of all rotamers that fall within 45 degrees of any of these three standard c1 values, F is the total number of rotamers in this distribution, and Ftot is the maximal number of rotamers obtainable for any distribution. The normalisation with respect to Ftot avoids that residues for which only very few position specific rotamers are found get a large value.
In the modeling process first a sparse model is generated. In this sparse model all conserved residues were left untouched, but other residues were mutated into alanine, unless they had to become glycine or proline. For all alanines that subsequently had to be mutated the rotameric entropy in this sparse model was determined. Side chains were than placed in order of decreasing rotameric entropy.
The sorting of residues as function of the rotameric entropy has an obvious advantage. Early in this modeling process the residues are built that have a very narrow rotamer distribution, which indicates that the conformation is mainly determined by the local backbone, and the absence of many not yet modeled residues is not a disadvantage. Residues with wider rotamer distributions which therefore are more influenzed by the rest of the molecule are built later when more residues are already completed. The advantage of this process is best seen from figure 14.
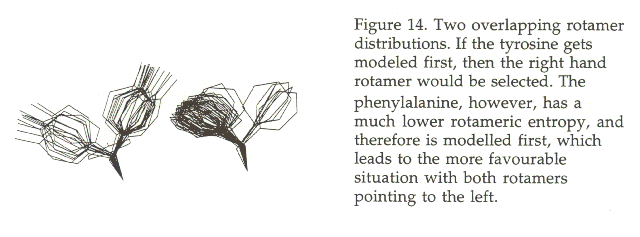
In practice, cases like the one described in figure 14 occur very often. Normally the number of rotamers left for side chains with many seemingly similar rotamer possibilities reduces strongly if the Śsimpleą residues are modelled first. For once, proteins are well behaved, and Murphy`s laws don`t apply.
|
|
It is obvious that database systems that allow for fast, easy and flexible retrieval of specific information are crucial for model building procedures. Several general [118-122] and single purpose [2,126] data storage or retrieval systems have been developed to extract information about protein sequences and structures from databases. Some of them hardly (re)organize data, but merely combine a database of three-dimensional protein structures with a set of algorithms for pattern recognition, data analysis and graphics. In general, these systems provide very flexible tools, but this flexibility is paid for by a rather low speed when the algorithms are applied to large amounts of data. PKB [118] and to some extent the parameter correlation method [119] are good examples of such systems. They are well suited for prototyping queries or searches in small subsets of the database, but less suitable for practical use if fast data extraction is required.
If retrieval times must be reduced to a minimum, one resorts to systems that pre-process and reorganize data to speed up the process of extracting information. Two important classes of such systems are object-oriented database systems (OODBS) and relational database systems (RDBS) [117]. An OODBS can easily search for many related objects, but the organization of the data makes it slow at doing sequential scans [121]. P/FDM [121] is a good example of an OODBS. Its high level query language Daplex is very concise and approaches the power of a programming language for complex queries.
In a protein RDBS many structural properties such as accessibility, torsion angles and secondary structure are stored in tables and queries are performed by logical combination of these tables. BIPED [120] and SESAM [122] are examples of such systems. SESAM does not fit the relational model exactly as it also provides some algorithms on top of the RDBS to allow for otherwise impossible or prohibitively slow queries. Advantages of a generalized RDBS [117] are the generally high speed of searches and the intuitive way in which queries are constructed.
When one wants to use a standard RDBS to aid with model building by homology one major problem is encountered: Entries in the same database table are assumed to be unrelated; or in other words, the database does not know which residues sit next to each other in the sequence. So, a query like 'buried - accessible - buried - accessible' to find surface b-strands is inherently beyond the capabilities of a standard relational system [120].
The SCAN3D database system [113] was specifically designed to bypass this problem. SCAN3D exploits the sorted character of protein structures in that it stores the residues or their characteristics in the database tables in the sequential order in which they occur in the protein. This allows to easily search for stretches of consecutive residues with specified characteristics, a feature that is especially important for the modeller because she is never interested in just one residue, but always in residues in their environment.
The Brookhaven Protein Databank (PDB) [123] contains atomic coordinates and some related information for more than 4000 macromolecular structures in plain text files. SCAN3D uses a representative set of slightly more than 300 protein structures [115,116]. These proteins are carefully selected to avoid bias towards a small number of abundantly present protein families. Therefore, results obtained from the use of SCAN3D queries are representative for the whole universe of presently known protein structures. Which allows this module of the WHAT IF program to also be used for basic studies of protein sequence-structure relatuons that later can lead to improved rules for homology modelling.
G.V. 9-May-1998