|
|
Rodriquez et al., [131] studied the structure of surface accessible loops because they realised that such loops are the major source of errors during modelling experiments. They studied 34 pairs of known structures and analysed what happened if they pretended not to know one of the two and then model it based on the other partner of the pair. Of course this is not fair because in the real world the structure of the model is never known, but from this study we can learn a lot about what can all go wrong upon modelling proteins. They concentrated on loops with equal length in the two structures to avoid having to model insertions and deletions and found three reasons why one often observes different conformations in similar structures:
Symmetry contact in model or in template 46 Symmetry contact in model and in template 43 No symmetry contacts in either of the two 27 Mutations involving proline 25 Mutations involving glycine 26 Mutations involving proline and glycine 4 Cases without any obvious reason for model problems 17 Symmetry contacts combined with proline or glycine 38Table 1. Most probable reason for the conformational differences between loops in homologous structures. In 75% of all cases crystal symmetry contacts are involved. Not all numbers add up correctly because multiple problems can occur at the same time. Only 17 out of 116 cases were not trivially put in any of the three major problem categories.
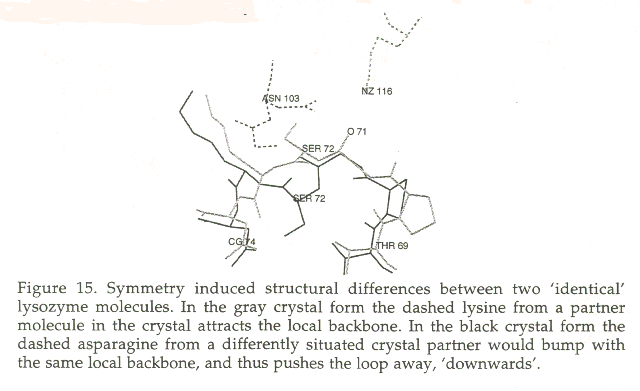
The largest fraction of all problems is clearly different symmetry contacts. This problem provides a principal limit to the accuracy of the model and to the posibilities of estimating the reliability of models. Figure 15 and 16 show two examples of symmetry induced conformational differences.
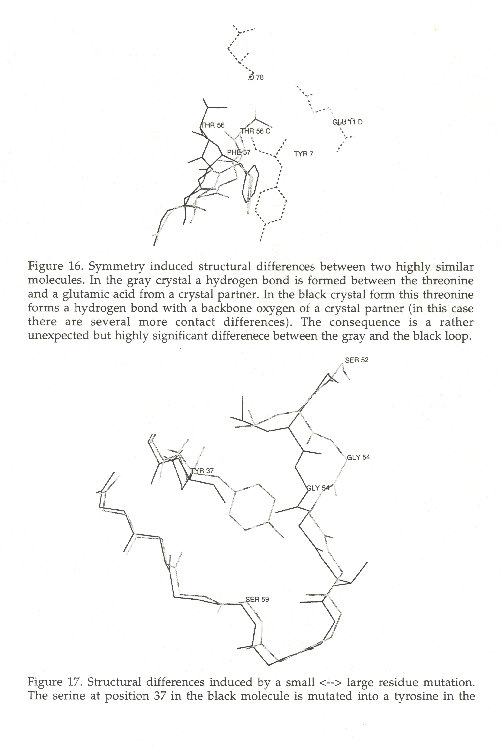
A more predictable scenario ocurrs when the backbone has to move to make space for a bulky residue as can be seen in figure 17 in which the 2.99 Angstrom displacement of the loop from residues 54 to 59 in 1POH [130] relative to 1PTF [132] is probably caused by the mutation of Ser37 in 1POH to Tyr in 1PTF.
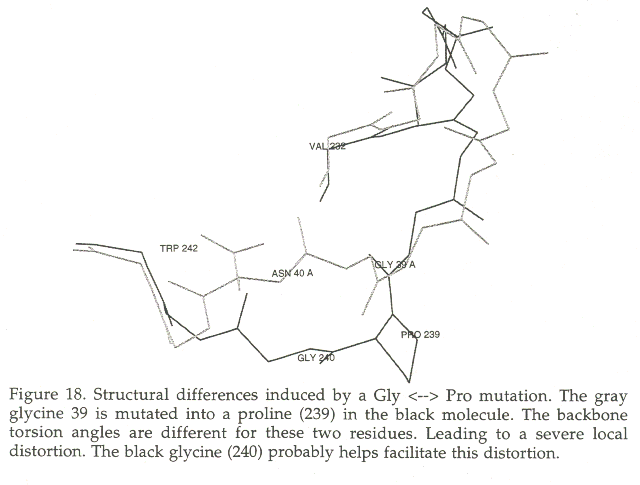
Modelling a proline is another big problem. When a proline replaces another residue in many cases the existing backbone has torsion angles that are very unfavourable for proline, and the proline insertion leads to local backbone adaptations. The worst cases are often found for the Gly->Pro mutation, because glycine can have almost every conformation without restrictions. Figure 18 shows the superposition of 5HPV [133] and 1IVP [134]. The loop from residues 34 to 42 is shown to illustrate the change in the backbone conformation due to the mutation of Gly to Pro at position 39 in 5HPV. The backbone at this position can not accomodate a proline (f=115.2, y=131.0, w=177.9). Proline 39 sits in 1IVP in a favourable conformation (for a Pro) (f=-87.5, y=137.9, w=179.6). The residues in the neighbourhood are are also influenced by these backbone torsion angle differences and the loop shows a maximum Ca-Ca displacement of 3.2 Angstrom for the Ca of residue 40.
|
|
In case of 75% or more sequence identity between the structure and the model one seldomly encounters insertions or deletions, and when they are encountered, they normally are short.
One of the major problems in model building with intermediate homology is the insertion of loops. If an insertion in the sequence occurs relative to the structure, there is no template to model on, and other techniques have to be applied. The techniques used to model loops are:
|
|
All models built by homology will have errors. Sidechains can be placed incorrectly, or whole loops can be misplaced. As with most errors, they become less of a problem when they can get localized. For example, upon modeling a protease it is probably not important that a loop far away from the active site is placed incorrectly.
The most important step in the process of model building by homology is therefore undoubtedly the verification of the model, and the estimation of the likelihood and magnitude of errors.
There are two principally different ways to estimate errors in a structure.
The key aspect is the development of criteria with sufficient discriminatory power to distinguish a good model from a bad one. An example is provided by deliberately misfolded proteins in which the sequence of a protein known to have an all-helical 3D structure is placed into a known structure of a completely different type, an antiparallel b- barrel, and vice versa. For the evaluation of the quality of these clearly incorrect hypothetical structures, intramolecular energy, calculated in vacuum using standard empirical potentials, is not a sensitive criterion [Novotny et al 84, 88]. The free energy difference between the folded and unfolded states would be an ideal criterion, but present theories are not capable of calculating free energy differences to sufficient accuracy.
Faced with the lack of an accurate theory of protein folding, empirical observations of regularities gleaned from the database of solved structures can be very useful. A variety of statistical criteria, which measure the preferential distribution of hydrophobic side chains in the interior of proteins, have been used successfully to discriminate between deliberately misfolded and native structures [64,149-151].
Normality indices for structures have already proven their power in structure verification. Many characteristics of protein structures lend themselves for normality analysis. Most of them are directly or indirectly based on the analysis of contacts, either inter residue contacts, or contacts with water. Some published examples are:
Atomic contacts are observed because they are energetically favored. Real structures cannot tolerate too many unfavorable interactions. Thus for a model to be correct only a few infrequently observed atomic contacts are allowed. We made a detailed analysis of atom atom contacts [155]. WHAT IF [135] holds a module that compares the local contact patterns with the average contact patterns for similar residue-residue contacts found in the database. This method can be summarized as follows: If a residue-residue contact has the same contact patterns and the same spatial orientation as a contact that occurs often in the database then a high score is given. If a contact in the modeled molecule seems rather unique, either from a point of view of which residues make the contact, or from a point of view of directionality of the contact, a low score is given. This 'quality control' of local packing has proven to be a powerful tool for the detection of abnormal structures. Most methods used for the verification of protein structures can also be used for the verification of models. Not all methods will be useful because certain experimental errors simply are not made by the better modelling programs. In general, however, a verification report is very helpful for the modeller and her friends when they are using the model for the analysis of experimental results or prediction of new experiments.
|
|
The quality of protein models built by homology to a template structure is normally determined by the RMS errors in models of proteins of which the structure is known. Rodriquez et al., selected from the PDB [ref] 34 pairs of protein structures that superpose well, have 35% to 98% sequence identity, and have no insertions or deletions. They created this test-set to analyze what could potentially be the major sources for errors in protein modelling and in the assessment of the model quality.
The dataset was carefully selected to be representative for the universe of proteins, but they made sure that they would not encounter big surprises. The models are thus representative for the best scenario one can expect in practical cases, and not for a typical scenario. The selection of 34 pairs of proteins was done using the following criteria:
PDB r R RMSd SID % Class RMSe Description 1poh 2.00 0.14 1.978 Phosphotransferase (E. coli) 1ptf 1.60 0.16 1.244 35.29 mixed 1.977 Phosphotransferase (S. faecalis) 1nhk 1.90 0.17 2.410 Nucleoside Diphosphate Kinase (M. xanthus) 1ndc 2.00 0.18 1.554 43.75 mixed 2.082 Nucleoside Diphosphate Kinase (D. discoideum) 1bpt 2.00 0.17 2.003 Pancreatic Trypsin Inhibitor (BPTI) (B. taurus) 1aap 1.50 0.18 0.973 44.64 mixed 1.984 PInh. Domain Of Alzheimer's Protein (H. sapiens) 5pal 1.54 0.17 1.626 Parvalbumin (T. semifasciata) 1omd 1.85 0.17 0.776 44.86 alpha 1.375 Oncomodulin (R. norvegicus) 1pza 1.80 0.18 1.752 Pseudoazurin (A. faecalis) 1pmy 1.50 0.20 0.995 45.00 beta 1.807 Pseudoazurin (M. extorquens) 1thbB 1.50 0.20 1.972 Hemoglobin (H. sapiens) 1pbxB 2.50 0.18 1.240 45.21 alpha 1.983 Hemoglobin (P. bernacchii) 5hvpB 2.00 0.18 1.716 HIV-1 Protease (HIV Type 1) 1ivpA 2.50 0.20 0.892 48.48 beta 1.531 HIV-2 Protease (HIV Type 2) 2sam 2.40 0.19 1.496 SIV-1 Protease (SIV Type 1) 4phvB 2.10 0.18 1.030 51.52 beta 1.863 HIV-1 Protease (HIV Type 1) 2cro 2.35 0.20 1.872 434 Cro Protein (Phage 434) 2or1L 2.50 0.18 0.825 52.38 alpha 1.882 434 Repressor (Phage 434) 1crb 2.10 0.19 1.423 Cellular Retinol Binding Protein (R. rattus) 1opbC 1.90 0.17 0.718 56.39 beta 1.436 Cellular Retinol Binding Protein II (R. rattus) 1fkf 1.70 0.17 1.287 FK-506 Binding Protein (H. sapiens) 1yat 2.50 0.18 0.818 57.01 beta 1.189 Fk-506 Binding Protein (S. cerevisiae) 1pvaA 1.65 0.20 1.244 Parvalbumin (E. lucius) 1cdp 1.60 0.16 0.702 62.04 alpha 1.130 Parvalbumin (C. carpio) 2ycc 1.90 0.20 1.390 Cytochrome C (S. cerevisiae) 5cytR 1.50 0.16 0.574 62.14 alpha 1.386 Cytochrome C (T. alalunga) 1azrA 2.40 0.17 1.469 Azurin (Pseudomonas aeruginosa) 1aizA 1.80 0.17 0.982 63.28 mixed 1.443 Azurin (Alcaligenes denitrificans) 4azuA 1.90 0.18 1.387 Azurin (Pseudomonas aeruginosa) 1azcA 1.80 0.16 0.960 63.78 mixed 1.332 Azurin (A. denitrificans) 1mrj 1.60 0.17 1.291 Alpha-trichosanthin (T. kirilowii maxim) 1mom 2.16 0.19 0.626 65.04 mixed 1.350 Momordin (M. charantia) 1cad 1.80 0.19 0.999 Rubredoxin (P. furiosus) 8rxnA 1.00 0.15 0.604 66.67 mixed 1.001 Rubredoxin (D. vulgaris) 1tadB 1.70 0.21 1.636 Transducin-alpha (B. taurus) 1gia 2.00 0.17 1.139 69.35 alpha 1.576 Gi Alpha 1 (R. rattus) 1hsaA 2.10 0.20 1.736 Human Class I HSA (H. sapiens) 1vaaA 2.30 0.17 1.176 72.63 mixed 1.829 MHC Class I (M. musculus) 1gbt 2.00 0.16 0.798 Beta-trypsin (B. taurus) 1brcE 2.50 0.17 0.424 73.09 beta 0.865 Trypsin Variant (R. rattus) 1babB 1.50 0.16 0.968 Hemoglobin Thionville (H. sapiens) 1fdhG 2.50 0.32 0.513 73.29 alpha 0.933 Hemoglobin (H. sapiens) 1dhfA 2.30 0.18 1.397 Dihydrofolate Reductase (H. sapiens) 1dr7 2.40 0.16 0.775 75.27 mixed 1.242 Dihydrofolate Reductase (G. gallus) 8dfr 1.70 0.19 1.335 Dihydrofolate Reductase (G. gallus) 2dhfA 2.30 0.19 0.738 75.27 mixed 1.456 Dihydrofolate Reductase (H. sapiens) 1hna 1.85 0.23 1.611 Glutathione S-transferase (H. sapiens) 3gstB 1.90 0.16 1.025 75.58 alpha 1.431 Glutathione S-transferase (R. rattus) 1ala 2.25 0.20 1.042 Annexin V (G. gallus) 1avr 2.30 0.18 0.445 77.85 alpha 0.882 Annexin V (H. sapiens) 1bra 2.20 0.16 0.999 Trypsin (R. rattus) 1mct 1.60 0.17 0.421 79.82 beta 1.044 Trypsin (S. scrofa) 4p2p 2.40 0.21 2.099 Phospholipase A2 (S. scrofa) 2bpp 1.80 0.19 1.152 84.17 alpha 1.922 Phospholipase A2 (B. taurus) 135l 1.30 0.19 1.213 Lysozyme (M. gallopavo) 1hhl 1.90 0.17 0.732 86.82 alpha 1.184 Lysozyme (N. meleagris) 2gbp 1.90 0.15 0.891 Galactose binding protein (E. coli) 3gbp 2.40 0.16 0.518 94.43 mixed 0.918 Galactose binding protein (S. typhimurium) 1emy 1.78 0.15 1.330 Myoglobin (E. maximus) 1ymc 2.00 0.13 0.691 87.58 alpha 1.324 Sulfmyoglobin (E. caballus) 1ovb 2.30 0.20 1.593 Ovotransferrin (Duck) 1nnt 2.30 0.16 1.091 90.57 mixed 1.572 Ovotransferrin (G. gallus) 2lalA 1.80 0.19 0.970 Lentil Lectin (L. culinaris) 2ltnA 1.70 0.18 0.322 92.27 beta 0.977 Pea Lectin (P. sativum) 2chf 1.80 0.18 1.955 Chey (S. typhimurium) 1chn 1.76 0.19 1.376 97.62 mixed 1.963 Chey (E. coli) 1etb1 1.70 0.16 0.678 Transthyretin (H. sapiens) 1ttcA 1.70 0.18 0.255 98.31 beta 0.534 Transthyretin mutant (H. sapiens)Table 2. Structures used to study model quality[135]. RMSd: Root mean square displacement between equivalenced atoms in the two molecules. RMSe: Root mean square atomic misplacement between the model and the real structure. SID: percentage sequence identity between a pair of sequences. R: crystallographic R-factor. r: resolution.
Additionally the dataset should be "representative" for the universe of globular water soluble protein structures that are amenable to modelling by homology. Roughly equally many all-alpha, all-beta and mixed alpha-beta proteins were chosen, and they were distributed equally over the 35-98% pairwise sequence identity range in all these three classes. Table 2 lists the pairs of proteins used, as well as some vital statistics.
Most modelling procedures use the backbone of the template as the backbone of the model, and add the sidechains onto this backbone. The RMSe of the backbone will therefore be the same as the RMSd between the model and template backbone. We call this the starting error. Obviously, under normal conditions the final all atom RMSe will always be bigger than this starting error. Energy based calculations are not yet refined enough to improve the results significantly (see next paragraph). Statistical methods can indicate "where" backbone modifications are likely to be needed, but except for some simple cases, we can not yet predict "how" to modify the backbone.
Loops normally have roughly a similar conformation in similar structures. A weak correlation is found between differences in loop conformations and mutations involving proline or glycine. However, if loops are not predicted well, this is most often the result of differences in symmetry contacts between these loops in the model and the template structure.There is a basic error of around 1.0 Angstrom in the backbone of every model, just as a result of differences between experimental structures. Surface located residues and structural changes caused by symmetry contacts add on average another 0.5 Angstrom to the RMS. In the core the error is normally much less than 1.0 Angstrom. At the surface itb is often more than 2.0 Angstrom. Of course some models will have lower RMS errors, but the problem is that in practical cases one cannot know how good the models are, one can only gamble [61].
|
|
All 68 models were energy minimised using GROMOS [156] (other programs give the same or similar results) and after a fixed number of energy minimisation steps the half minimised structures were evaluated. The RMSe was measured, and all 68 RMSe values measured after 100, 200, etc., energy minimisation steps were bluntly averaged. The results are summarised in table 3. Two things are clearly seen. 1) The improvements than can be achieved are minimal, and 2) The energy minimisation run should be short, after a while the models get worse again. Table 3 is only an average, but inspection of all individual numbers shows that the optimum is in all but three cases between 50 and 300 energy minimisation steps. Inspection of some individual energy minimisation processes indicates that during the first steps the largest errors (such as two atoms being a bit to close to each other, or a hydrogen bond that does not have optimal geometry, or a backbone angle that was already not perfect in the template, etc.) are removed. At every step, however many, many very small errors are introduced. In the beginning removal of the big errors outweighs the introduction of the many small errors. If after a while all larger problems are solved, the only thing that still happens is the introduction of many small errors.
Steps Ave. RSMe 0 1.4622 100 1.4542 200 1.4529 300 1.4335 500 1.4552 2000 1.4553 8000 1.4553Table 3. Average RMSe after a fixed number of energy minimisation steps. The average RMSe was calculated averaging the RMSe of the 68 individual structures in each of the energy minimisation runs.
|
|
Most of the above deals with modeling in three dimensions. That is, it is assumed that a good model can be built. Other techniques such as secondary structure prediction can help in this case. It is often not clear why predicted secondary structures are at all published, but in the hands of a biocomputing expert some information can be extracted from the prediction. The best secondary structure prediction program that is available (this is written on august 17 1996) is without doubt PHD. This program can be used via the WWW (see below).
Future developements in protein modelling are the use of other information than homology to build models. Such information can essentially be anything. Predicted secondary structure, accessibility or contacts can equally well be used as observed cysteine bridges, proteolytic cleavage sites or accessibilities.
|
|
Model building by homology is a young field. Many improvements can still be made and much work still needs to be done to make these improvements. Our modeller can still learn a lot from the professional gambler, but we expect that improvements in energy calculation based software will within 10 years lead to a breakthrough. We would not be surprised if untill this happens improving the odds of present day methods by inclusion of information from multiple templates, the design of new algorithms and heuristics, better and larger databases, the rapid growth of the PDB, and a few more factors that we cannot yet predict, will step by step create the progress in homology modelling that is needed to close the structure gap.
WWW addresses Secondary structure prediction: http://swift.embl-heidelberg.de/predictprotein/ Protein structure quality: http://swift.embl-heidelberg.de/pdbreport/ http://biotech.embl-heidelberg.de:8400/ Protein structure comparison: http://www.ebi.ac.uk/dali/
|
|
We thank Chris sander, Rob Hooft, Glay Chinea, Enzo de Filippis,
Hans Doeberling and his team, Brigitte Altenberg, Karina Krmoian for
stimulating discussions and practical help. We appologise to the people
working on other good modelling programs (especially Ruben Abagyan
and Andrej Sali) for not having enough space to explain their methods
and programs in detail. We appologise to the numerous
crystallographers who made all this work possible by depositing
structures in the PDB for not referring to each of the 4000 very important
articles describing these structures.
This article was written by R.Rodriguez and G.Vriend.
|
|
1) The relation between the divergence of sequence and structure in proteins. Chothia, C., Lesk, A.M., EMBO J., 5 (1986) 823-836. 2) Database of homology-derived protein structures and the structural meaning of sequence alignment. Sander, C., Schneider, R., PROTEINS, 9 (1991) 56-68. 3) Modelling by homology. Swindells, M.B., Thornton, J.M., Curr.Op.Struct.Biol., 1 (1991) 219-223. 4) Structural relationships of homologous proteins as a fundamental principle in homology modeling. Hilbert, M., B�hm, G., Jaenicke, R., PROTEINS, (1993), 17, 138-151. 5) How different amino acid sequences determine similar protein structures: the structure and evolutionary dynamics of the globins. Lesk, A.M., Chothia, C., J.Mol.Biol., (1980) 136, 225-270. 6) On the use of sequence homologies to predict protein structure: identical pentapeptides can have completely different conformations. Kabsch, W., Sander, C., PNAS, (1984) 81, 1075-1078. 7) Evolution of proteins formed by b-sheets. I. Plastocyanin and Azurin. Chothia, C., Lesk, A.M., J.Mol.Biol., (1982) 160, 309-323. 8) Knowledge-based model building of proteins: concepts and examples. Bajorath, J., Stenkamp, R., Aruffo, A., Prot.Sci., (1993) 2, 1798-1810. 9) Homology modelling: inferences from tables of aligned sequences. Lesk, A.M., Boswell, D.R., Cuur.Op.Struc.Biol.. (1992) 2, 242-247. 10) A new method for building protein conformations from sequence alignments with homologues of known structure. Havel, T.F., Snow, M.E., J.Mol.Biol., (1991) 217, 1-7. 11) Rebuilding flavodoxin from Ca coordinates: a test study. Reid, L.S., Thornton, J.M., PROTEINS, (1989) 5, 170-182. 12) Comparative modeling of homologous proteins. Greer, J., Meth.Enzym., (1991) 202, 239-252. 13) Homology modeling of divergent proteins. Sudarsanam, S., March, C.J., Srinivasan, S., J.Mol.Biol., (1994) 241, 143-149. 14) Protein model building using structural homology. Lee, R.H., Nature, (1992) 356, 543-544. 15) Comparative modelling by satisfaction of spatial restraints. Sali, A., Blundell, T.L., (1993) 234, 779-815. 16) Modelling of globular proteins. A distance based search procedure for the construction of insertion regions and pro <--> non-pro mutations. Summers, N.L., Karplus, M., J.Mol.Biol., (1990) 216,991-1016. 17) Prediction of homologous protein structures based on conformational searches and energetics. Schiffer, C.A., Caldwell, J.W., Kollmann, P.A., Stroud, R.M., PROTEINS, (1990) 8, 30-43. 18) Modelling by homology. Swindells, M.B., Thornton, J.M., Curr,Op.Struc.Biol., (1991) 1, 219-223. 19) A large scale experiment to assess protein structure prediction methods. Moult, J., Pedersen, J.T., Judson, R., Fidelis, K., PROTEINS, (1995) 23, 2-4. 20) A critical assessment of comparative molecular modeling of tertiary structures of proteins. Mosimann, S., Meleshko, R., James, N.G., PROTEINS, (1995) 23, 301-317. 21) Analysis of six protein structures predicted by comparative modelling techniques. Harrison, R.W., Chatterjee, D., Weber, I.T., Proteins, (1995) 23, 463- 471. 22) Homology modelling by the ICM method. Cardozo, T., Totrov, M., Abagyan, R., PROTEINS, (1995) 23, 403-414. 23) Homology modelling of histidine-containing phosphocarrier protein and eosinophil-derived neurotoxin: construction of models and comparison with experiment. Church, W.B., Palmer, A., Wathey, J.C., Kitson, D.H., PROTEINS, (1995) 23, 422-430. 24) Confronting the problem of interconnected structural changes in the comparative modeling of proteins. Samudrala, R., Pedersen, J.T., Zhou, H.-B., Luo, R., Fidelis, K., Moult, J., PROTEINS, (1995) 23, 327-336. 25) Evaluation of comparative protein modeling by MODELLER. Sali, A., Potterton, L., Yuan, F., Vlijmen, H. van, Karplus, M., PROTEINS, (1995) 23, 318- 326. 26) Modelling mutations and homologous proteins. Sali, A., Curr.Op.Struc.Biol., (1995) 6, 437-451. 27) Detection of common three dimensional substructures in proteins. Vriend, G., Sander, C., PROTEINS (1991) 11, 52-58. 28) Multiple protein structure alignment from tertiary structure comparison: assignment of global and residue confidence levels. Russell, R.B., Barton, G.J., PROTEINS (1992) 14, 309-323. 29) Identification of protein folds: Matching hydrophobicity patterns of sequence sets with solvent accessibility patterns of known structures. Bowie, J.U., Clarke, N.D., Pabo, C.O., Sauer, R.T., PROTEINS (1990) 7, 257-264. 30) Identification of tertiary structure resemblance in proteins using a maximal common subgraph isomorphism algorithm. Grindley, H.M., Artymiuk, P.J., Rice, D.W., Willett, P., J.Mol.Biol., (1993) 229, 707-721. 31) The alignment of protein structures in three dimensions. Zuker, M., Somorjai, R.L., Bull. Math.Biol. (1989) 51, 55-78. 31) A rapid method for protein structure alignment. Orengo, C.A., Taylor, W.R., J.Theor.Biol., (1990) 147, 517-551. 32) Comparison of three-dimensional structures of homologous proteins. Overington, J.P., Curr.Op.Struc.Biol., (1992) 2, 394-401. 33) A variable gap penalty function and feature weights for protein 3-D structure comparisons. Zhu, Z.-Y., Sali, A., Blundell, T.L., Prot.Engin., (1992) 5, 43-51. 34) Fast structure alignment for database searching. Orengo, C.A., Brown, N.P., Taylor, W.R., PROTEINS (1992) 14, 139-167. 35) Size independent comparison of protein three dimensional structures. Maiorov, V.N., Crippen, G.M., PROTEINS, (1995) 22, 273-283. 36) Common spatial arrangements of backbone fragments in homologous and non-homologous proteins. Alexandrov, N.N., Takahashi, K., Go, N., J.Mol.Biol., (1992) 225, 5-9. 37) An efficient automated computer vision based technique for detection of three dimensional structural motifs in proteins. Fisher, D., Bachar, O., Nussinov, R., Wolfson, H., J.Biolol.Struct.&Dyn., (1992) 9, 769-789. 38) Techniques for the calculation of three dimensional structural similarity using inter-atomic distances. Pepperrell, C., Willett, P., J.Comp.-Aid.Mol.Des., (1991) 5, 455-474. 39) Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. Maiorov, V.N., Crippen, G.M., J.Mol.Biol., (1994) 235, 625-634. 40) A protein structure comparison methodology. Brown, N.P., Orengo, C.A., Taylor, W.R., Comp.Chem. (1996) 20, 359-380. 41) Protein structure alignment. Taylor, W.R., Orengo, C.A., J.Mol.Biol., (1988) 208, 1-22. 42) Definition of general topological equivalence in protein structures. Sali, A., Blundell, T.L., J.Mol.Biol., (1990) 212, 403-428. 43) Comparison of conformational characteristics in structurally similar protein pairs. Flores, T.P., Orengo, C.A., Moss, D.S., Thornton, J.M., Prot.Sci., (1993) 2, 1811-1826. 44) Protein structure comparison by alignment of distance matrices. Holm, L., Sander, C., J.Mol.Biol., (1993) 233, 123-138. 45) Biological meaning, statistical significance, and classification of local spatial similarities in nonhomologous proteins. Prot.Sci., (1994) 3, 866-875. 46) Founding fathers and families. Br�nd�n, C.-I., Nature, (1990) 346, 607-608. 47) A database of protein structure families with common folding motifs. Holm, L., Ouzounis, C., Sander, C., Tuparev, G., Vriend, G., Prot.Sci., (1992) 1, 1691- 1698. 48) Searching protein structure databases has come of age. Holm, L., Sander, C., PROTEINS, (1994) 19, 165-173. 49) SCOP: A structural classification of proteins database for investigation of sequence and structures. Murzin, A.G., Brenner, S.E., Hubbard, T., Chothia, C., J.Mol.Biol., (1995) 247, 536-540. 50) OB (oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. Murzin, A.G., EMBO, (1993) 12, 861-867. 51) Structural features can be unconserved in proteins with similar folds. Russell, R.B., Barton, G.J., J.Mol.Biol., (1994) 244, 332-350. 52) Different protein sequences can give rise to highly similar folds through different stabilizing interactions. Laurents, D.V., Subbiah, S., Levitt, M., Prot.Sci., (1994) 3, 1938-1944. 53) Thiol proteases. Comparative studies on the high resolution structures of papain and actinidin, and on amino acid sequence information for cathepsins B and H, and stem bromelian. Kamphuis, I.G., Drenth, J., Baker, E.N., (1985) 182, 317-329. 54) Similarity of active-site structures. Pearl, L., Nature, (1993) 362, 24. 55) Three-dimensional, sequence order-independent structural comparison of a serine protease against the crystallographic database reveals active site similarities: potential implications to evolution and to protein folding. Fisher, D., Wolfson, H., Lin, S.L., Nussinov, R., Prot.Sci., (1994) 3, 769-778. 56) Plastic adaptation toward mutation in proteins: structural comparison of thymidilate synthases. Perry, K.M., Fauman, E.B., Finer-Moore, J.S., Montfort, W.R., Maley, G.F., Maley, F., Stroud, R.M., PROTEINS, (1990) 8, 315-333. 57) Three dimensional structural resemblance between leucine aminopeptidase and carboxypeptidase A revealed by graph-theoretical techniques. Artymiuk, P.J., Grindley, H.M., Park, J.E., Rice, D.W., Willett, P., FEBS Lt., (1992) 303, 48-52. 58) Recurrence of a binding motif? Swindells, M.B., Orengo, C.A., Jones, D.T. Pearl, L.H., Thornton, J.M., Nature, (1993) 362, 299. 59) PROCHECK: a program to check the stereochemical quality of protein structures. Laskowski, R.A., MacArthur, M.W., Moss, D.S., Thornton, J.M., J.Appl.Cryst., (1993) 26, 283-291. 60) Stereochemical quality of protein-structure coordinates. Morris, A.L., MacArthur, M.W., Hutchinson, E.G., Thornton, J.M., PROTEINS, (1992) 12, 345-364. 61) Errors in protein structures. Hooft, R.W.W., Vriend, G., Sander, C., Abola, E.E., Nature, (1996) 381, 272. 62) Recognition of errors in three dimensional structures of proteins. Sippl, M.J., PROTEINS, (1993) 17, 355-362. 63) Assessment of protein models with three dimensional profiles. L�thy, R., Bowie, J.U., Eisenberg, D., Nature, (1992) 356, 83-85. 64) Criteria that discriminate between native proteins and incorrectly folded models. Novotny, J., Rashin, A.A., Brucoleri, R.E., PROTEINS, (1988) 4, 19-30. 65) Knowledge-based prediction of protein structures and the design of novel molecules. Blundell, T.L., Sibanda, B.L., Sternberg, M.J.E., Thornton, J.M., Nature, (1987) 326, 347-352. 66) Amino acid pair interchanges at spatially conserved locations. Naor, D., Fisher, D., Jernigan, R.L., Wolfson, H.J., Nussinov, R., J.Mol.Biol., (1996) 256, 924-938. 67) Environment-specific amino acid substitution tables: tertiary templates and prediction of protein folds. Overington, J., Donnelly, D., Johnson, M.S., Sali, A., Blundell, T.L., Prot.Sci., (1992) 1, 216-226. 68) Recognition of distantly related proteins through energy calculations. Abagyan, R., Frishman, D., Argos, O., PROTEINS, (1994) 19, 132-140. 69) An empirical energy function for threading protein sequence through the folding motif. Bryant, S.H., Lawrence, C.E., PROTEINS, (1993) 16, 92-112. 70) Prediction of protein structure by evaluation os sequence structure fitness. Ouzounis, C., Sander, C., Scharf, M., Schneider, R., J.Mol.Biol., (1993) 232, 805- 825. 71) Threading a database of protein cores. Madej, T., Gibrat, J.-F., Bryant, S.H., PROTEINS, (1995) 23, 356-369. 72) Protein structure prediction by threading methods: evaluation of current techniques. Lemer, C.M.-R., Rooman, M.J., Wodak, S.J., PROTEINS, (1995) 23, 337-355. 73) Fold recognition and ab initio structure predictions using hiddem markov models and b-strand pair potentials. Hubbard, T.J., Park, J., PROTEINS, (1995) 23, 398-402. 74) A branch-and-bound algorithm for optimal protein threading with pairwise (contactpotential) amino acid interactions. Lathrop, R.H., Smith, T.F., Proc. 27- th Hawaii Intl. Conf. on System Sciences (1994) IEEE Comp. Soc. Press. 365-374. 75) A structural basis for sequence comparisons. Johnson, M.S., Overington, J.P., J.Mol.Biol., (1993) 233, 716-738. 76) Structural analysis based on state-space modeling. Stultz, C.M., White, J.V., Smith, T.F., Prot.Sci., (1993) 2, 305-314. 77) A Method to identify protein sequences that fold into a known three dimensional structure. Bowie, J.U., L�thy, R., Eisenberg, D., Science, (1991) 253, 164-170. 78) Rapid and sensitive comparison with FASTA and FASTP. Pearson, W.R., Meth.Enzym., (1990) 183, 63-98. 79) Basic local alignment search tool. Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J., J.Mol.Biol., (1990) 215, 403-410. 80) Atomic environment energies in proteins defined from statistics of accessible and contact surface areas. Delarue, M., Koehl, P., J.Mol.Biol., (1995) 249, 675-690. 81) Evaluation of protein models by atomic solvation preference. Holm, L., Sander, C., J.Mol.Biol., (1992) 225, 93-105. 82) Identification of protein sequence homology by consensus template alignment. Taylor, W.R., J.Mol.Biol., (1986) 188, 233-258. 83) A fast and sensitive multiple sequence alignment algoritm. Vingron, M., Argos, O., CABIOS, (1989) 5, 115-121. 84) A method for multiple sequence alignment with gaps. Subbiah, S., Harrison, S.C., J.Mol.Biol., (1989) 209, 539-548. 85) Improving the sensitivity of the sequence profile method. L�thy, R., Xenarios, I., Bucher, P., Prot.Sci., (1994) 3, 139-146. 86) Pattern-induced multi sequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for use in comparative protein modelling. Smith, R.F., Smith, T.F., Prot.Engin., (1992) 5, 35-41. 87) Sequence ordinations: a multivariate analysis approach to analysing large sequence data sets. Higgins, D.G., CABIOS, (1992) 8, 15-22. 88) A strategy for the rapid multiple alignment of protein sequences. Confidence levels from tertiary structure comparisons. Barton, G.J., Sternberg, M.J.E., J.Mol.Biol., (1987) 198, 327-337. 89) Recognition of related proteins by iterative template refinement. Yi, T.-M., Lander, E.S., Prot.Sci., (1994) 3, 1315-1328. 90) The three dimensional profile method using residue preference as a continuous function of residue environment. Zhang, K.Y.J., Eisenberg, D., Prot.Sci., (1994) 3, 687-695. 91) A possible three-dimensional structure of bovine a-lactalbumin based on that of hen�s egg-white lysozyme. Brown, W.J., North, A.C.T., Phillips, D.C., Brew, K., Vanaman, T.C., Hill, R.C., J.Mol.Biol., (1969) 42, 65-86. 92) Computation of structure of homologous proteins: a-lactalbumin from lysozyme. Warme, P.K., Momany, F.A., Rumball, S.V., Scheraga, H.A., Biochemistry (1974) 13, 768-782. 93) Prediction of protein side-chain conformations from local three dimensional homology reletionships. Laughton, C.A., J.Mol.Biol., (1994) 235, 1088-1097. 94) Analysis of the relationship between side-chain conformation and secondary structure in globular proteins. McGregor, M.J., Islam, S.A., Sternberg, M.J.E., J.Mol.Biol., (1987) 198, 295-310. 95) Tertiary templates for proteins. Ponder, J.W., Richards, F.M., J.Mol.Biol., (1987) 193, 775-791. 96) Rotamers, to be or not to be? Schrauber, H., Eisenhaber, F., Argos, O., J.Mol.Biol., (1993) 230, 592-612. 97) Fast and simple Monte Carlo algorithm for side chain optimization in proteins: application to model building by homology. Holm, L., Sander, C., PROTEINS, (1992) 14, 213-223. 98) Modelling of side chains, loops and insertions in proteins. Summers, N.L., Karplus, M., Meth.Enzym., (1991) 202, 156-205 99) Construction of side-chains in homology modelling. Application to the C- terminal lobe of rhizopuspepsin. Summers, N.L., Karplus, M., J.Mol.Biol., (1989) 210, 785-811. 100) A method to configure protein side-chains from the main-chain trace in homology modelling. Eisenmenger, F., Argos, O., Abagyan, R., (J.Mol.Biol., (1993) 231, 849-860. 101) The dead-end elimination theorem and its use in protein side-chain positioning. Desmet, J., Maeyer, M. De., Hazes, B., Lasters, I., Nature, (1992) 356, 539-542. 102) New paths from death ends. Taylor, W., Nature, (1992) 356, 478-480. 103) Predicting local structural changes that result from point mutations. Filippis, V.de, Sander, C., Vriend, G., Prot.Engin., (1994) 7, 1203-1208. 104) Backbone-dependent rotamer library for proteins. Application to side-chain prediction. Dunbrack, R.L.Jr., Karplus, M., J.Mol.Biol., (1993) 230, 543-574. 105) Evidence for strained interactions between side-chains and the polypeptide backbone. Stites, W.E., Meeker, A.K., Shortle, D., J.Mol.Biol., (1994) 235, 27-32. 106) Conformational analysis of the backbone dependent rotamer preferences of protein side chains. Dunbrack, R.L.Jr., Karplus, Nature Struc.Biol., (1994) 5, 334- 340. 107) The use of position specific rotamers in model building by homology. Chinea, G., Padron, G., Hooft, R.W.W., Sander, C., Vriend, G., PROTEINS, (1995) 23, 415-421. 108) Detailed ab initio prediction of lysozyme-antibody complex with 1.6 A accuracy. Totrov, M.M., Abagyan, R.A., Nature Struct. Biol., (1994) 1, 259-265. 109)Accurate prediction of stability and activity effects of site directed mutagenesis on a protein core. Lee, C., Levitt, M., Nature (1991) 352, 448-451. 110) Prediction of the stability and activity effects of site directed mutagenesis. Gunsteren, W.F. van, Mark, A.E., J.Mol.Biol., (1992) 227, 389-395. 111) Thermodynamics of protein peptide interactions in the ribonuclease S system studied by molecular dynamics and free energy calculations. Simonson, T., Brunger, A.T., Biochemistry (1992) 31, 8661-8674. 112) Prediction and analysis of structure, stability and unfolding of thermolysin like proteases. Vriend, G., Eijsink, V.G.H., J.Comp.-Aid Mol.Des. (1993) 7, 367- 396. 113) A novel search method for protein sequence-structure relations using property profiles. Vriend, G., Sander, C., Stouten, P.W.F., Prot.Engin. (1994) 7, 23-29. 114) Using known substructures in protein model building and crystallography. Jones, T.A., Thirup, S., EMBO, J., (1986) 5, 819-823. 115) Selection of representative protein data sets. Hobohm, U., Scharf, M., Schneider, R., Sander, C., Prot.Sci., (1992) 1, 409-417. 116) Verification of protein structures: side-chain planarity. Hooft, R.W.W., Sander., C., Vriend, G., Cabios, accepted. 117) Intelligent databases. Parsaye K., Chignell, M., Khoshafian, S., Wong, H., John Wiley and sons, Inc., (1989). 118) PKB: A program system and data base for analysis of protein structure. Bryant, S.H., PROTEINS (1989) 5, 233-247. 119) Parameter relation rows: a query system for protein structure function relationships. Vriend, G., Prot.Engin., (1990) 4, 221-223. 120) A relational data base of protein structures designed for flexible enquiries about conformation. Prot.Engin., (1989) 2, 431-442. 121) An object oriented database for protein structure analysis. Gray, P.M.D., Paton, N.W., Kemp, G.J.L., Fothergill, J.E., Prot.Engin., (1990) 3, 235-243. 122) SESAM: A relational database for structure and sequence of macromolecules. Huysmans, M., Richelle, J., Wodak, S.J., PROTEINS, (1991) 11, 59-76. 123) The protein data bank: A computer based archival file for macromolecular structures. Bernstein, F. C., Koetzle, T. F., Williams, G. B., Meyer, E. F. Jr.,Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T. ; Tatsumi, M. J.Mol.Biol. (1977) 112, 535-542. 124) IPSA-Inductive protein structure analysis. Schultze-Kremer, S., King, R.D., Prot.Engin., (1992) 5, 377-390. 125) GBPARSE: a parser for the GenBank flat-file format with new feature table format. Read, R.L., Davison, D., Chappelear, J.E., Garavelli, J.S., CABIOS, (1992) 8, 407-408. 126) A cross reference table between the protein data bank of macromolecular structures and the national biomedical research foundation protein identification resource amino acid sequence data bank. Lesk, A.M., Boswell, D,R., Lesk, V.I, Lesk, V.E., Bairoch, A., Prot.Seq.Data.Anal., (1989) 2, 295-308. 127) The EMBL data library. Stoehr, P.J., Cameron, G.N., NAR, (1991) 19, 2227- 2230. 128) Protein motifs and database searching. Thorton, J.M., Gardner, S.P., TIBS, (1989) 14, 300-304. 129) A profile for molecular biology databases and information resources. Kamel, N.N., CABIOS, (1992) 8, 311-321. 130) To be published. Jia, Z., Quail, J. W., Waygood, E. B., Delbaerre L. T. J. (1993), Deposited in the PDB. 131) Limits to modelbuilding by homology. Rodriguez, R., Vriend, G., to be submitted. 132) To be published. Jia, Z., Vandonselaar, M., Hengstenberg W., ,Quail, J. W., Delbaerre L. T. J. (1993), Deposited in the PDB. 133) Crystallographic analysis of a complex between human immunodeficiency virus type 1 protease and acetyl pepstatin at 2.0 Angstrom resolution. Fitzgerald, P. M. D., Mc Keever, B. M., Van Middlesworth, J. F., Springer, J. P., Heimbach, J. C., Leu, C. T., Herber, W. K., Dixon, R. A. F., Darke, P. L. (1990) J. Biol. Chem. 265, 14209-. 134) Refined 1.6 A resolution crystal structure of the complex formed between porcine b-trypsin and MCTI-A, a trypsin inhibitor of the squash family. Huang, Q., Liu, S., Tang, Y. (1993) J. Mol. Biol. 229, 1022-. 135) WHAT IF: A molecular modelling and drug design program. G. Vriend, J.Mol.Graph. (1990) 8, 52-56. 136) Hubbard, R.E., In: Computer Graphics and molecular modelling. Edt. Fletterick, R.J., Zoller, M., Cold Spring Harbor, (1986) 9-12. 137) A graphics modelbuilding and refinement system for macromolecules. Jones, T.A., J.Appl.Cryst. (1978) 268-272. 138) Interactive program for visualization and modelling of proteins, nucleic acids and small molecules. Dayringer, H.E., Tramontano, A., Fletterick, R.J., J.Mol.Graph. (1986) 4, 82-87. 139) Improved methods for buildin protein models in electron density maps and the location of errors in these models. Jones, T.A., Zou, J.Y., Cowan, S.W., Kjelgaard, M., Acta Cryst A (1991) 47, 110-119. 140) BRAGI: A comprehensive protein modelling program system. Schomburg, D., Reichelt, J., J.Mol.Graph. (1988) 6, 161-165. 141) An algorithm for determining the conformation of polypeptide segments in proteins by systematic search. Moult, J., James, M.N.G., PROTEINS (1986) 1, 146-163. 142) Prediction of the folding of short polypeptide segments by uniform conformational sampling. Bruccoleri, R.E., Karplus, M., Biopolymers (1987) 26, 137-168. 143) Predicting antibody hypervariable loop conformations. II: minimization and molecular dynamics studies of MCPC603 from many randomly generated loop conformations. Fine, R.M., Wang, H., Shenkin, P.S., Yarmush, D.L., Levinthal, C., PROTEINS (1986) 1, 342-362. 144) A new method for building protein conformations from sequence alignments with homologues with know structure. Havel, T.F., Snow, M.E., J.Mol.Biol. (1990) 217, 1-7. 145) Assembly of polypeptide and backbone conformations from low energy ensambles of short fragments. Sippl, M.J., Hendlich, M., Lackner, P., Prot.Sci. (1992) 1, 625-640. 146) Calculation of protein conformation as an assembly of stable overlapping segments: application to BPTI. Simon, I., Glasser, L., Scheraga, H.A., PNAS (1991) 88, 3661-3665. 147) On the multiple minima problem in the conformational analysis of polypeptides. Ripoll, D.R., Scheraga, H.A., Biopolymers (1990) 30, 165-176. 148) A large scale experiment to assess protein structure prediction methods. Moult, J., Judson, R., Fidelis, K., Pedersen, J.T., PROTEINS (1995) 23, ii-iv. 149) Polarity as a criterion in protein design. Baumann, G., Froemmel, C., Sander, C., Prot.Engin. (1989) 2, 329-334. 150) Correctly folded proteins make twice as many hydrophobic contacts. Bryant, S.H., Amzel., L.M., Int.J.Pept.Prot.Res. (1987) 29, 46-52. 151) Identification of native protein folds amongst a large number of incorrect models. Hendlich, M., Lackner, P., Weitcus, S., Floeckner, H., Froschauer, R., Gottsbacher, K., Cassari, G., Sippl, M.J., J.Mol.Biol. (1990) 216, 167-180. 152) Stereochemical quality of protein structure coordinates. Morris, A.L., MacArthur, M.W., Hutchinson, E.G., Thorton, J.M., PROTEINS (1992) 12, 3456- 364. 153) Solvation energy in protein folding and binding. Eisenberg, D., McLachlan, A.D., Nature, (1986) 319, 199-203. 154) Novel method for the rapid evaluation of packing in protein structures. Gregoret, L.M., Cohen, F.E., J.Mol.Biol. (1990) 211, 959-974. 155) Quality control of protein models: directional atomic contact analysis. Vriend, G., Sander, C., J.Appl.Cryst. (1993) 26, 47-60. 156) GROMOS. Van Gunsteren, W.F., Berendsen, H.J., (1987) BIOMOS, Biomolecular software, Lab. Phys. Chem., Uni., Groningen, The Netherlands.
© June 21 2000 G Vriend